
Though the new European Union Medical Device Regulation (MDR) is set to fully apply from May 2021, there’s still uncertainty around what the new requirements will mean in practice.
Of particular interest is Rule 11, which is set to tighten requirements for Software as a Medical Device (SaMD), and, consequently, has the potential to lead to a broad swath of reclassifications for existing SaMD devices.
This blog explains the key things you need to know about Rule 11 and the broader MDR requirements, as well as the impact it’s likely to have for medical companies and other operators in the space.
What does MDR Rule 11 actually say – and what does it mean for medical device companies?
Under Rule 11 of the MDR, pretty much any SaMD that provides clinical information – such as information used for making decisions for diagnosis or treatment – will be classified as a Class IIa medical device or higher.
Under the existing Medical Device Directive (MDD) rules, the majority of this type of SaMD would have been Class I, the lowest risk category.
This ‘up-classification’ means medical companies creating digital health solutions have a range of new rules and requirements to comply with.
What exactly are the new MDR classification rules and regulations?
Many companies may be unfamiliar with the extent of EU medical device regulation, but they’ll need to be aware of what their new path to market could look like.
One important aspect is the conformity assessment procedure required for MDR certification, which, for most SaMD providers, will require the involvement of a Notified Body. At present, however, there are concerns around the limited availability of these Notified Bodies – currently there are just 16 listed in the European Commission’s New Approach Notified and Designated Organisations (NANDO) database.
Companies will also have to provide a sufficient level of clinical evidence to demonstrate conformity with the MDR general safety and performance requirements (which may well be more significant than the evidence required in the past).
Beyond this, there’s also the requirement to manage both pre-market and post-market cybersecurity risks, as well as a need to adhere to requirements for other economic operators in the supply chain, like manufacturers, distributors, and importers.
What does this mean for medical device classification in the meantime?
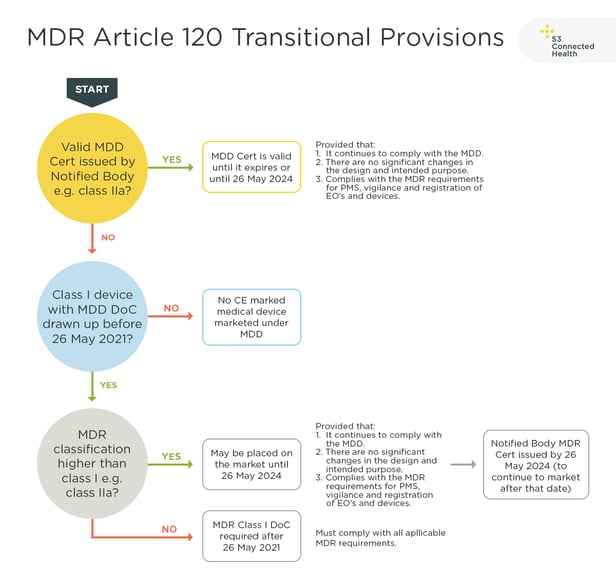
Some of the medical companies we work with are currently availing of the transitional arrangements, provided under MDR Article 120 (3).
This allows devices certified under MDD and on the market prior to the 26 May 2021 deadline can remain on the market until 26 May 2024, by which time they will need to be resubmitted under MDR. However, there are conditions that must be met.
For an MDD Class I device to avail of the transition it must be classified as Class IIa or higher under the new MDR classification rules. If the MDD device is still classified as Class I under the MDR rules, it cannot avail of the transition. However, most MDD Class I SaMD will be MDR Class IIa or higher under MDR Rule 11.
So, if you have an MDD Class I device and you want to continue marketing it after 26 May 2021, you have two choices:
1) CE mark the device under the new MDR before 26 May 2021
2) Avail of the MDR Article 120 (3) transition and continue to market the device as an MDD Class I device until 2024. After 2024 it will need to be CE marked under the MDR to remain on the market.
Remaining on the market until 2024 also requires that post-market surveillance and vigilance comply with MDR requirements, too, and that no significant changes be made to the device.
For any company looking to use the transitional arrangements, they’ll need to ensure that the restriction on making ‘significant changes’ to their apps during the transition period aligns with their overall product strategy.
What are the pros and cons of the new medical device classification rules?
Rule 11 will make the path to market for most SaMD more challenging, for example, with increased timelines and costs. This could impact smaller companies and startups, in particular. On the other hand, the eventual impact of the MDR will be to further protect and enhance public health by ensuring the safety and performance of SaMD throughout their lifecycle.
In many cases however, the impact is impossible to predict. It will take time to find the right balance between patient safety and timely access to innovative digital health products, and, initially, I expect there will be fewer medical devices and digital health apps coming to the EU market. The MDR is a significant shift, and it will take some time for both regulators and the industry to assess if that balance is right.
Whatever happens, it makes sense to be aware of the changes and what ‘up-classification’ means. Other digital health technology developments – like AI and machine learning, for example – will require further adaptation of the regulations and related guidance, too, and digital health solution manufacturers will need device approval and monitoring methods that better fit the agile development methodologies used in SaMD development.
Our team of regulatory experts help ensure your devices and SaMD meet all the necessary legal requirements. If you’d like to know more about how the upcoming EU MDR changes will affect you, and how to prepare, please get in touch.
Padraig Maguire
Head of Quality and Regulatory Affairs
S3 Connected Health